

- Address
- 浙江省宁波市高新区聚贤路587弄15号2#楼033幢12-1-13
- Phone
- 13367268046
- 854062773@qq.com



XRD的基本原理与应用
1 引言
XRD(X-ray Diffraction)中文全称是X射线衍射,是一种快速、准确、高效的材料无损检测技术。作为一种表征晶体结构及其变化规律的手段,其应用遍及材料、化学、生物、医药、陶瓷、冶金、矿产等诸多领域。但大多数新同学对于XRD的测试原理一知半解,对其应用停留在简单的物相鉴别阶段,对其不同样品的测试要求和注意事项也不甚清楚。在此,我们用最简洁易懂的语言对XRD从原理到应用进行了详细的总结,希望能给大家一些帮助。
2 XRD的测试原理
2.1 X射线是什么?
1895年,德国物理学家W.K.伦琴首次发现了X射线的存在,故X射线又称伦琴射线。X射线的本质是一种波长极短(约为10-8 ~10-12 m),能量很大的电磁波,它具有波粒二象性。[1] 现代科学研究中,对于波长<0.1 nm的X射线称为硬X射线,主要用于材料探伤;对于波长介于0.25~0.05 nm的称为软X射线,一般用于晶体结构分析。
2.2 X射线在晶体中的衍射行为
由于X射线具有极大的能量,当其到晶体中时,晶体中的原子会在X射线的作用下被迫做周期性的运动,从而会以原子球为单位对外发射次生波,该波的频率与入射X射线一致,这个过程就成为X射线的散射。考虑到晶体中的原子在空间上呈周期性的规律排布,这些散射球面波之间存在着固定的位相关系,会在空间产生干涉,结果导致在某些散射方向的球面波相互加强,而在某些方向上相互抵消,从而也就出现衍射现象。[2] 因此,晶体中的X射线衍射实质上就是大量原子散射波在空间上相互干涉的结果。
2.3 X射线衍射与材料结构的关联
对于非晶体材料,由于其结构不存在晶体结构中原子排列的长程有序,只是在几个原子范围内存在着短程有序,故非晶体材料的XRD图谱为一些漫散射馒头峰。然而,对于晶体材料,其原子排布在三维空间上长程有序,其XRD衍射图谱只在特定的位置上出现加强峰(X射线衍射加强结果)。晶体产生的衍射花样都反映出晶体内部的原子分布规律。
概括地讲,一个衍射花样的特征,可以认为由两个方面的内容组成:
a) 衍射线在空间的分布规律—由晶胞的大小、形状和位向决定;
b) 衍射线束的强度—取决于原子的种类和它们在晶胞中的位置。这些衍射花纹就像晶体的指纹一样,通过鉴别衍射花纹的在空间上出现的位置和强度,即可在X射线衍射和晶体结构之间建立定性和定量关系。
(1)布拉格方程是衍射分析中最重要的基础公式,是XRD理论的基石。它简单明确地阐明衍射的基本内涵,揭示了衍射与晶体结构的内在关系。如图3所示,当X射线照射到晶体中时,X射线在照射到相邻两晶面的光程差是2dsinθ。如果光程差等于X射线波长的n倍时,X射线的衍射强度将相互加强,反之在其他地方衍射强度不变或减弱。
nλ=2dsinθ, (n=1, 2, 3…..)
其中,λ, d, θ分别代表了X射线的波长,晶体晶面间距,入射X射线与相应晶面的夹角。
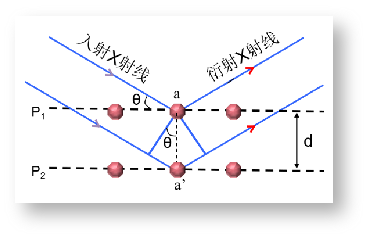
图3 X射线在晶体中多个原子面的反射情况
显然,通过布拉格方程,可以用已知波长的X射线去求解晶体晶面间距d,从而获得晶体结构信息,这就是结构分析;也可以用已知晶面间距的晶体来测量未知X射线的波长,这就是X射线光谱学。
(2)谢乐公式(Scherrer公式)则是XRD测晶粒度的理论基础。它主要描述了晶粒尺寸与衍射峰半峰宽之间的关系。晶粒越小,XRD衍射线的峰就越弥散宽化;反之则越集中。
D=Kλ/Bcosθ
其中D, K, λ, B, θ分别为晶粒垂直于晶面方向的平均厚度、Scherrer常数、X射线波长、实测样品衍射峰半高宽度(弧度)以及衍射角。Scherrer常数K的值一般由B来决定,当B为衍射峰半高宽,K=0.89;当K为衍射峰面积积分半高宽时,K=1。由于材料中的晶粒大小并不完全一样,故该方法计算的是不同大小晶粒的平均尺寸。
3 XRD衍射仪
3.1 仪器介绍
布拉格实验装置(图4)是现代X射线衍射仪的原型。如图4所示,XRD衍射仪的核心部件是X光源发生器和X射线检测器。当入射X射线照射到样品表面后,在满足衍射定律的方向上设置X光检测器,同时记录强度和衍射角θ(即入射线和反射面的夹角)。为了保证X光检测装置始终处于反射线的位置,X光检测装置和样品台必须始终保持以2:1的角速度同步转动。由此可见,发生X衍射的晶面始终是与试样表面平行的晶面。
需要说明的是,由于X发生器产生的光源是含有大量波长不一的X光(Kα, Kβ, 连续谱),如果这些波都参与衍射,得到的衍射峰将会杂乱无章。此外,在单一的X射线照射样品表面时也有可能激发出样品的特征射线,影响测试结果。因此,现代的X射线衍射仪为了保证测量的精度,往往还会在样品和X射线检测器之间加装单色器或滤波器,以获得优质的衍射图样。
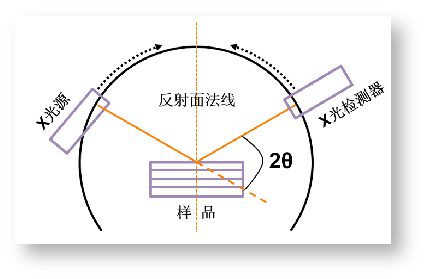
图4 布拉格实验装置简图
3.2 仪器使用注意事项
相对于仪器的使用和样品的制备,大部分同学可能更关注XRD测试时测量方式和实验参数的选择。首先是X射线的选择,X射线发生器的靶材对X射线的波长影响最大,常用的靶材有Cu、Co、Fe、Cr、Mo、W (表1)。由于某些靶材产生的X射线能使部分样品产生强的荧光吸收,因此选择合适的靶材是获得优质数据的第一步。最常用的Cu靶几乎适用于除含Cu和Fe的所有试样,稳定性高兼容性好;Co、Fe靶则分别适宜用单色器(Co)/滤波器(Fe)来测试Fe系样品;Cr靶也具有优秀的兼容性,能测试大部分试样,包括含铁样;Mo靶由于波长短适合奥氏体的定量分析;W靶具有连续X射线强的特点,常用于单晶的劳厄照相。
其次是测量参数的选择,包括了测定方式、扫描速率、扫描范围。测定方式分为连续扫描和步进扫描两种,前者适合于定性分析和微量检测,后者则适用于计算晶胞参数、结晶度、分析微应变、以及Rietveld精修。[1-3] 扫描速率的一般范围微0.001°-8°/min,同样地,根据测试需求选择不同的扫速,1°-8°/min适合定性和一般定量,0.001°-1°/min则适合定量计算。XRD测试的范围一般在2°-150°之间,定性分析一般取2°-90°,微量检测、定量分析以及点阵参数计算要保证待测样主衍射区完整,结晶度以及Rietveld精修一般在2°-150°之间测量。[2-4]
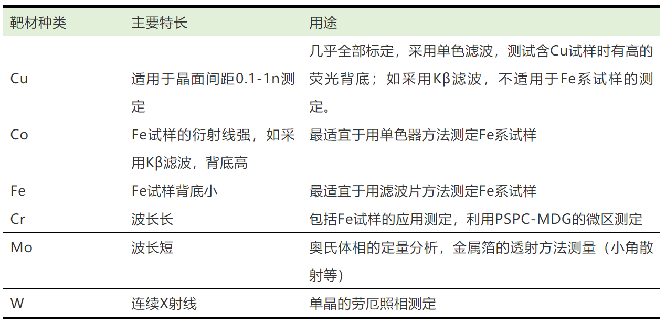
表1 常见靶材的种类和用途[1]
4 XRD的典型应用
XRD的典型应用可以分为定性和定量两部分,常用的XRD分析有以下五大类(图6):(1)物相定性;(2)确定晶胞参数;(3)晶体取向度分析;(4)晶粒尺寸计算;(5)物相定量计算。下面我们结合实际案例对这些应用逐一进行详解。

图6 XRD的典型应用
4.1 物相定性分析
物相分析是XRD测试中最常见的应用,每一相的衍射图形是唯一的,就像每个人的指纹一样,只要我们将测得的XRD图谱和数据库中的标准卡片进行对比,即可确定其结构。根据XRD谱图与标准谱的对比,我们可以得出以下信息:
i) 样品是无定形还是晶体, 无定型样品为大包峰,没有精细谱峰结构;晶体则有丰富的谱线特征;
ii) 所测样品的物相组成,纯相还是非纯相;
iii) 判断晶胞是否膨胀或者收缩。
如图7所示,待测样XRD峰形尖锐突出,与辉沸矿ZnSe标准卡高度重合,无明显的杂质峰存在,因此可判定待测样为为高纯的ZnSe。图8给出了同类物质不同结晶态的衍射图谱。不难在图8a中只有1个强峰和1个弱峰,这说明该材料内晶粒存在明显的取向,因此判断样品可能是单晶;而在图8b中,样品在所有的标准位置都检测到明显的衍射峰,说明该材料内晶粒存取向是各向异性的,因此判断样品可能是多晶。图9给出了Se掺杂的硫化锌样品在不同掺杂量下的XRD图谱,可以看出随着Se掺杂量的增加,样品的衍射峰出现的位置逐渐向左偏移。结合布拉格衍射方程,可知衍射角变小,说明晶面间距变大。
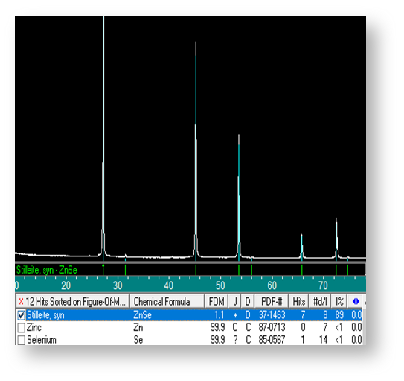
图7 ZnSe的XRD图谱
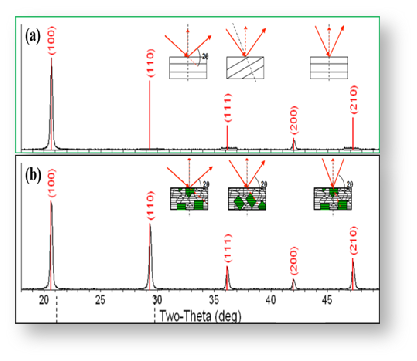
图8 同一物质单晶和多晶的XRD图谱
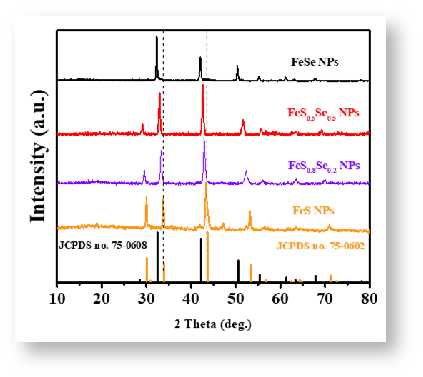
图9 FeS在不同Se掺杂量下的XRD图谱
4.2 定量计算
除了定性分析,XRD也能定量计算。常见的定量计算应用有:
a) 利用谢乐公式计算样品的平均晶粒尺寸;
b) 样品的相对结晶度;
c) 利用Rietveld全谱精修测定点阵常数、分析应力应变、获得键长键角信息等;
d) 利用K值法或Rietveld全谱精修定量确定不同物相在晶体中的含量。
在陶瓷材料和结构材料中,晶粒尺寸对其性能影响较大。晶粒尺寸的计算依据是谢乐公式:当X射线入射到小晶体时,其衍射线条将变得弥散而宽化,晶体的晶粒越小,X射线衍射谱带的宽化程度就越大。因此我们可以通过测定确定样品衍射峰的半高宽来计算晶粒大小。
材料的结晶度对材料性能的影响尤为显著。测定结晶度的方法很多,但不论哪种方法都是根据结晶相的衍射图谱面积与非晶相图谱面积决定,即结晶度=(衍射峰强度/总强度)×100%。
尽管不通过精修就能获得样品的结构参数,但是获得的结果往往是偏离实际的。因此为了提高结构参数的精度,采用Rietveld全谱拟合的方法对晶体结构的修正,从而得到材料的准确结构信息是很有必要的。
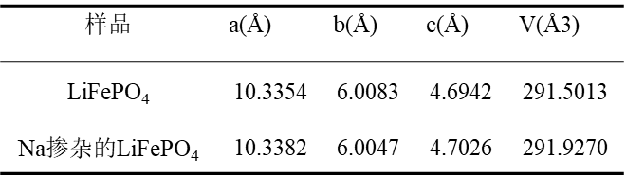
表2 精修后样品的晶胞参数[5]
XRD的测试及分析贯穿整个材料领域的研究,其在认识材料结构,解释材料性能,指导科学研究和实际生产等方面具有非常重要的作用。掌握并能有效利用XRD技术是每一个材料人必备的傍身技能。在此,推荐几本XRD分析的相关书籍供大家参考,希望各位有所收获。